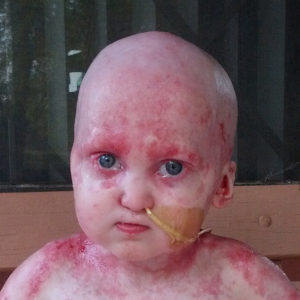
Ankyloblepharon-ectoderm defects-cleft lip and/or palate (AEC) syndrome is a type of ectodermal dysplasia characterized by severe skin erosion, cleft lip and palate, and fused eyelids. (In the past, AEC syndrome was also known as Hay-Wells syndrome or Rapp Hodgkin syndrome. We now understand they are the same condition and call it AEC syndrome.) Skin erosions can appear at birth or anytime during infancy. They are often very severe, can reoccur and last for a long time, resulting in bleeding and infections. No specific therapies are currently available for skin erosion. AEC and other similar ectodermal dysplasias are caused by mutations in the TP63 (also known as p63) gene.

The National Foundation for Ectodermal Dysplasias (NFED) has provided research funding to the labs of both Dr. Maranke Koster and Dr. Caterina Missero. We granted $25,000 to Dr. Koster in 2017. Both of these labs are interested in understanding the molecular defects that lead to skin erosions in individuals affected by AEC syndrome. The two groups are using different approaches to identify the underlying mechanisms that lead to cell and tissue fragility in the presence of TP63 mutations. Identifying these abnormalities will ultimately be necessary for designing therapies aimed at alleviating the skin erosion in AEC patients.
The work these researchers are doing is very complex. We acknowledge that it may be tough to understand all of the science in these updates. But, we are excited about the work they are doing to work towards therapies that would eliminate skin erosion once and for all!
Ectodermal dysplasia can cause a lifetime of challenges. By supporting research, you expand early diagnostics, treatments, pathways toward cures… and hope!Shape Our Futures With Research

AEC and EEC Research Update
By Maranke I. Koster and Peter J. Koch
University of Colorado Denver
The overall goal of our research program is to understand the molecular defects that lead to skin and cornea fragility in patients affected by AEC or ectrodactyly-ectodemal dysplasias-clefting (EEC) syndrome. As a first step towards this goal, we have collected skin biopsies from affected individuals. These biopsies are absolutely essential to understand the biological processes that lead to AEC or EEC.
Besides carrying the disease causing mutations in the TP63 gene, we believe that these cells also carry other genes that determine onset and severity of the disease. Consequently, only patient cells will enable us to completely understand these diseases. We used patient cells isolated from these biopsies to generate induced pluripotent stem cells (iPSC), a unique type of immortal stem cell that can generate all cell types of the body, including skin and eye cells, cell types affected in AEC and/or EEC patients.
Next, we used advanced gene editing tools to fix the disease-causing TP63 mutation in these cells (gene-corrected cells). We then differentiated patient-derived and gene-corrected stem cells into keratinocytes, the cells that make up the epidermis.
Our initial experiments focused on cells from AEC patients. When we compared the AEC keratinocytes to the gene-corrected keratinocytes, we found that many components of desmosomes, cell structures that connect keratinocytes with each other like “Velcro”, were not functioning properly. The inability of the patient cells to form stable cell-cell connections might explain why AEC patients often present with severe skin fragility. We are now performing additional experiments to further define the precise molecular defects in patient desmosomes, and to determine if we can restore cell adhesion in keratinocyte sheets derived from AEC patients.
In addition, we are performing similar experiments with EEC patient-derived cells to determine if AEC and EEC cells are similar and/or distinct disease mechanisms. Finally, we are also generating models to investigate the mechanisms underlying the corneal fragility and limbal stem cell deficiency that occur in many EEC patients. For this project, we are using both EEC patient-derived induced pluripotent stem cells and genetically engineered mice as disease models.

RESEARCH ON AEC SYNDROME AND SKIN EROSION
By Caterina Missero, Ph.D.
Center for Genetic Engineering and Advanced Biotechnology, and University of Naples Federico II, Italy
Some years ago, I met individuals and families affected by AEC syndrome at the AEC conferences organized by the NFED. It was a touching experience and I was struck by the fact that so little could be done for children with severe skin erosions. Since then, I became interested in understanding the molecular mechanisms underlying this disease. Our research group set up to study this disease at the molecular level with the long term goal of identifying therapies that would alleviate or possibly cure the severe skin erosions observed in children.
Previous studies on human epidermal cells had not been very useful to understand the disease due to its complexity. To model the disease in a complete organism, our research group has developed mouse models carrying a mutation in the p63 gene identical to one found in patients. The p63 protein is crucial for the skin because it regulates the expression of many proteins, and for this reason when the gene is mutated and the protein is not fully functional, many aspects of the normal physiology of the skin are affected.
We found that the most important function that is partially impaired in AEC syndrome is mechanical resistance. Being in contact with the outside world, the skin is a tissue subjected to mechanical stress and has to be flexible and resistant. To these purposes, the skin makes proteins with specialized functions, such as the keratins (structural filaments inside the cells) and desmosomes (strong connections between cells anchored with keratins). We found that in AEC syndrome the number of filaments and connections are reduced, and therefore the skin easily breaks and the most external layers are peeled off.
How do we restore mechanical resistance?
It is not simple to reconstitute filaments and connections, but if we could understand what goes wrong in p63 when it is mutated, we could think of ways to fix it. We noticed that p63 mutations causative of AEC syndrome are mainly found in a specific region of the gene with an unknown function and some of them have the characteristic of making a longer protein, otherwise absent in unaffected individuals.
To make sense of these observations, we started a very fruitful collaboration with Dr. Volker Dotsch, University of Frankfurt – Germany, an expert in p63 biochemistry. In a study recently published in the Proceedings of the National Academy of Sciences USA, we demonstrated that p63 mutants causative of AEC syndrome, but not those causative of the other related syndromes, disrupt p63 function by causing protein misfolding (incorrect structural 3D conformation of the protein) and aggregation with itself and possibly with other proteins. The protein is trapped in large non-functional complexes and therefore cannot perform its normal function. Protein aggregates were found not only in the mouse model but also in keratinocytes isolated from human patients.
Protein aggregation is typical of many other pathologies such as neurodegenerative diseases including Alzheimer’s and Parkinson’s diseases. In these cases, protein aggregates accumulate in the brain to a point that they become toxic to the cells. In AEC syndrome, protein aggregates are not as toxic, leading to the hope there may be ways to disentangle the p63 protein. Using a genetic approach, we demonstrated that p63 function is restored by reverting its aggregation.
These studies pave the way to discover new therapies for preventing accumulation of misfolded protein or reducing misfolding by stimulating cells to get rid of the aggregates (cellular clearance), thereby restoring normal function. We will use a combination of studies in human cells derived from patients and in mouse models using different approaches to design these new therapies for this untreatable disease.
This is so interesting. Is possible that the same treatment could help patients affected by Incontinentia Pigmenti, to heal the typical blisters they have in the skin? Or being from a different mutation would make this non viable? Thanks.
Hi, Adriana. You raise an interesting question. I’ll post it to the researcher and see what she says. Stay tuned. Jodi, NFED, Director of Marketing and Communication
Could this research help children with focal dermal hypoplasia?
Hi, Deana. For now, the researcher is only focusing on p63 conditions, not focal dermal hypoplasia. That said, it’s possible that if they learn something that works for the p63 conditions, they could investigate if a similar treatment is possible for focal dermal hypoplasia. But, at this time, we don’t know that it would help or work.
Hi,
My son has ED, he has a gene mutation on P63 gene. My daughter in law is 13 weeks pregnant. They went for high level sonogram yesterday. They are having a girl and she has both hands affected. Is there any way they can test further? Are there any studies or clinical trails that they can get involved in?
Thank you so much.
Janet Gruber
Hi, Janet:
Unfortunately, I am not aware of any clinical trials currently happening for the syndromes caused by the P63 mutation. We do have some information about prior studies on our website, which can be read here: https://nfed.org/research/research-studies/p63-syndrome-studies/.
If the doctor is seeing that her hands are affected (assuming your son has EEC?), that is one way of diagnosing in utero. Some of the syndromes can also be confirmed genetically, with in utero genetic testing. Your son and daughter-in-law would need to consult with a geneticist or genetic counselor to learn if this is beneficial or not, and if it is possible, pending the syndrome. You can read more about diagnosing babies, here: https://nfed.org/blog/diagnosing-ectodermal-dysplasia-syndromes-babies/
We would love to welcome your family to our community, and if you need additional resources for your granddaughter, please reach out to our Director of Family and Community programs, Kelley, at kelley@nfed.org.
Best of luck to you and your family!
Veronica