

In December of 2016, we shared with you some preliminary data from Dr. Holm Schneider’s study of treating three babies with x-linked hypohidrotic ectodermal dysplasia (XLHED) in utero. Today, we have an extraordinary update that gives us great cause for celebration! Dr. Schneider and his team of investigators have published their groundbreaking research results in The New England Journal of Medicine in an article titled “Prenatal Correction of X-Linked Hypohidrotic Ectodermal Dysplasia.” We are thrilled to share with you key highlights from their research, what it means for our families affected by XLHED, and the next steps.
What Causes XLHED?
The symptoms of XLHED are caused by a change in the EDA gene. The altered gene does not produce a protein called ectodysplasin A1 (EDA1). This is the protein that signals the normal growth of hair, teeth, skin and certain glands, like sweat and mucous glands, in the body. Research, partially funded by the National Foundation for Ectodermal Dysplasias (NFED), developed a synthetic version of EDA1.
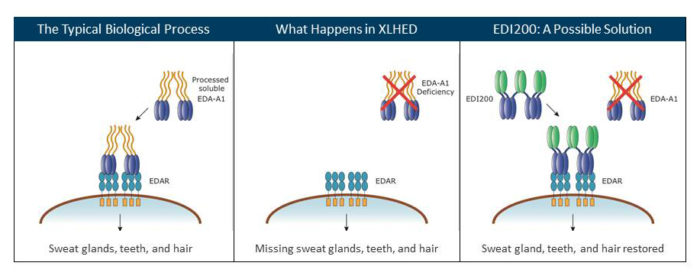
Scientists used this synthetic version called EDI200 to treat XLHED in mice, dogs and then finally infants in the Newborn XLHED Clinical Trial. The latter study conducted by Edimer Pharmaceuticals found that administering EDI200 to infants between days 2 and 14 of their lives was not significantly effective in correcting the symptoms. Because XLHED is a developmental disorder, treating after birth was too late in the process. Dr. Schneider and the other investigators hypothesized that studies were needed to administer the EDI200 at the developmental stage at which the sweat glands and other affected body parts start to develop in the fetus. This meant treating in utero.
The Latest Study
In 2016, two families in Germany asked and were considered for treatment by Dr. Schneider’s team. The mom of the first family suspected the twin sons she was carrying might be affected by XLHED. (Corinna is pictured above with the twins.) She also had an older son who has the condition.

First, the mom was confirmed to be a carrier of XLHED. Second, the German researchers used ultrasound technology to determine that she was carrying twin boys who were both lacking tooth buds. The ultrasound showed that neither had any tooth buds in the lower jaw. One boy had one tooth bud in the upper jaw and the other had two tooth buds. An MRI confirmed these findings and the diagnosis of XLHED for both boys. The mother received injections of EDI200 into the amniotic cavities for each fetus at 26 and 31 weeks.
For the second family, Dr. Schneider administered EDI200 to their son in utero at 26 weeks by amniocentesis. Again, ultrasound had shown lack of tooth buds. This child only received the one treatment because of a limited supply of EDI200.
The Findings
Overall, the findings are very positive!
Sweat Glands
How well the EDI200 restored the sweat glands was a key benchmark in determining if the treatment was successful. The news is phenomenal. The twins have a normal number of sweat glands. And the sweat glands are also functioning normally!
They produced amounts of sweat similar to infants who are not affected by XLHED. This is in great contrast to their older brother who does not sweat at all. The third baby who only had one treatment had slightly less sweat glands than an unaffected infant.

It’s extraordinary to think about how this lifelong challenge is removed for them and their parents. No longer do they need to worry about overheating and carrying cooling supplies. The fact that these boys can sweat normally is a huge treatment breakthrough!
Tooth Buds
The twins each had 10 and eight tooth buds compared to their untreated brother, age 5, who has three teeth and one tooth bud. It appears that the boys will not have the normal number of teeth. However, they do have a greater number, which is very positive. More teeth will allow better growth of their jawbone. This should improve their bite force, decrease the likelihood of needing a bone graft and help them sustain dental implants down the road, if needed.
Respiratory
At the time of the report, the twins were 22 months and had not had any respiratory-related hospitalizations. Many of our little ones with XLHED often suffer from pneumonias, respiratory infections and hospitalizations. Eliminating those issues is definitely a treatment success!
Salivary Glands
The twins produced a normal amount of saliva. This is great as restoring saliva will help with digesting food in the mouth, protecting enamel on teeth and with swallowing.
Meibomian Glands
The twins had more meibomian glands in their lower eyelids than their untreated brother. These are glands on the rim of the eyelid that produce an oily substance which helps prevent tear fluid from evaporating. Having an increased number of meibomian glands should help prevent dry eye problems.
Hair
EDI200 did not have any effect on hair. Given the early time point at which hair develops in the human fetus, this was not a surprise.
Relaunching The Clinical Trial
These findings provide us with great hope that the prenatal treatment does correct many of the symptoms for XLHED. However, we understand that this was a small study that involved only three babies. The next step is to conduct a study on a larger number of babies.
![]()
We are excited to report good news on this front as well! The EspeRare Foundation announced today that it is relaunching a clinical trial to establish the in utero protein therapy of XLHED as a safe, viable and effective treatment. Based in Switzerland, EspeRare is a nonprofit organization whose mission is to overcome roadblocks that prevent new therapies to reach rare diseases patients.
Caroline Kant, Founder and CEO of EspeRare, said that they have entered into an agreement with Edimer Pharmaceuticals for EspeRare to receive the full rights to continue the development of an innovative therapy for XLHED. EspeRare has renamed EDI200 as ER-OO4.
“This is such an exciting time for the EspeRare team who is preparing to relaunch the clinical development of ER-004 in the coming months, bringing back hope and aiming to provide very promising outcomes for the XLHED community. With this program, not only does EspeRare have the opportunity to bring to patients the first ever therapy that can address a genetic disease during pregnancy, but it also has the power to shift the rare diseases drug development paradigm, opening a new pathway to diagnose and correct other genetic diseases before birth.
EspeRare plans to launch the trial in the first half of 2019 with the ultimate goal of bringing this treatment to market! According to EspeRare, “these efforts will benefit from the European Medicines Agency’s (EMA) PRIME (PRIority MEdicines) scheme, due to the rarity of the disease, the absence of alternative treatment options and the encouraging results obtained in babies treated prenatally by Prof. Holm Schneider.”
Essentially, this means that the EMA will provide enhanced support to the clinical trial. Our Board of Directors, Scientific Advisory Council (SAC) and staff are committed to continue working with EspeRare and offering support as needed.
Dr. J. Timothy Wright, a member of our SAC, has helped steer our XLHED research program for decades.
“Since discovery of the cause of XLHED in 1996, there has been an ongoing effort to find a potential treatment,” Dr. Wright said. “Development of a fusion protein to replace the non-functional protein in the early 2000s showed great promise when used to treat mice and dogs having genetic alterations similar to humans. Since that time, there have been a series of human studies indicating the treatment is safe. This most recent report is the first to describe an intrauterine therapy to replace the protein from a genetic abnormality and demonstrates the tremendous promise for treatment of XLHED using this approach. It is extremely exciting.
According to EspeRare, ER-004 is the first and only treatment specifically targeting XLHED. Administered at the right time during fetal development, it has the potential to become a “single course” treatment, effectively switching off symptoms of the disorder throughout patients’ lives.
Thanking Those Who Paved the Way
Dr. Schneider will continue to monitor the three babies his team has treated. We, the NFED, have awarded $75,000 to him to fund this important work.
We are enormously grateful to Dr. Schneider and his investigative team for being tenacious in their efforts to give our families affected by XLHED a treatment to help correct their symptoms. We salute the two families who stepped forward to be a part of this ground breaking study. Their willingness to participate helped us confirm what we learned in the mice models research: that indeed, the prenatal treatment of XLHED with ER-004is a viable treatment.
They are not the only families who are the heroes in this story. Rather, this research success rests on the shoulders of thousands of XLHED families who have participated in studies for the past 30+ years. Families who literally gave blood, hair and skin samples, who completed surveys, and who volunteered their infants for the phase II clinical trial. They did their part in the faith that a future generation might benefit from a treatment. And that treatment is closer than ever before.
Sarah Yaroch, who participated in the Newborn XLHED Clinical Trial, said Dr. Schneider’s results make her hopeful.
“I’m happy these three won’t have to struggle as much. We participated in the clinical trial knowing our son might not benefit. We did it to help the research. If we had anything to do with this new success, we are thankful we could help. We are thankful to the people before us who volunteered for research, the adults who were treated first to be sure the treatment was safe. There are so many players who helped us get to where the research is right now. Everybody took a part!”
We especially thank all of the donors who supported the NFED for three decades to be the driving force in XLHED research. You can share in our pride in this achievement we have accomplished together! We hope that we can continue to count on you. Consider supporting our Impact Cures, Now! Campaign to raise funds for this critical research for XLHED by donating.
And of course, we must thank our founder, Mary K. Richter, for having the vision for our research program. Her decades of leadership were paramount in getting the research world interested in this condition.
The journey to this point has been up and down but we have always moved forward with optimism and unwavering commitment. The path moving forward will likely be the same. We know that we still have many years to go and successes to be had before this treatment is available for families. But, we will keep moving forward!
For today, join us in celebrating this research milestone and the hope it provides our ectodermal dysplasias community!
Ectodermal dysplasia can cause a lifetime of challenges. By supporting research, you expand early diagnostics, treatments, pathways toward cures… and hope!Shape Our Futures With Research
You can also watch and listen to my announcement on this Facebook Live video.
https://www.facebook.com/NationalFoundationforEctodermalDysplasias/videos/10155349729971716/
Not everyday ,but I used to thought what the solution for this ,y my child are suffering so lot …..when I read a article Abt proprioceptive passive expansion appliance there I found miracle and today unimaginable one …it’s really really great ….God is here among us only ……. sir ur work cannot be described by means of word ….but it’s really excellent ….
How the thanks be given for your wonderful gift. But we are very eagerly waiting for your announcement for cure of who are suffering from xlhed adults.
You are welcome. We understand your desire for a treatment to help children and adults who are affected by XLHED. We wish for that, too! Unfortunately, this treatment only works to correct the symptoms when it’s given to a person before those affected body parts have developed. This means giving the treatment in utero, while the baby is still in the mother’s womb. Maybe some day, researchers will be able to develop such a treatment. However, this treatment works only when given in utero. Jodi, NFED Director, Marketing and Communications
I am very happy to hear about the cure of XLHED …
Hi, Amna. Just to clarify, this treatment is not cure in that the person treated would still have the gene and has a chance of passing it to a child. However, it does correct the symptoms! Thanks, Jodi, NFED Director, Marketing and Communications
This is such exciting news. Kudos to all who had a part in this. To make life easier for a child is all we can ask for. Truly wonderful
Great news and a great article. Can you comment on plans for bringing this treatment to the United States?
EspeRare has long range plans to expand the clinical trial to the United States down the road. We don’t have any specifics at this time. Jodi, Director, NFED Marketing and Communications
This is amazing! I actually started to cry while reading this story because decisions around having children burdened me for years.
My husband and I decided on IVF with PGD to have our son in 2015, he does not have XLHED. I am so glad the soon down the road there will be more options for people like me who want to have children but bear the burden, struggling with what are the right options. Most insurance doesn’t cover IVF or PGD so that makes it very difficult financially to take that path. It also is a difficult moral or ethical decision for some.
I hope that this continuing research can find a balance of when and how often to provide the ER-004 for best results for the child! I will certainly be watching for updates on the progress and trials.
Thank you for the comments. From what we understand, the investigators plan to administer the ER-004 two times, at weeks 26 and 31. Jodi, NFED Director, Marketing and Communications
Is a treatment for adults close?
Unfortunately, this treatment was found to only work in babies in utero. The treatment is administered before or at the time t he body part is developing. The treatment was tested on babies after they were born and the results were not significant. So, this treatment would not help adults as their body parts are fully formed. Unfortunately, we are not aware of any treatment for adults at this time. But, research keeps progressing, so who knows what could happen some day. Jodi, NFED, Director, Marketing and Communication
Maybe stem cell research for others. just to be able to sweat would be a life changer.
Hi, I am currently 14 weeks pregnant. I have a healthy 2 year old son, but I myself have ED and was just told by my doctor that the baby I am carrying might have a cleft palate, which for me was a symptom of ED. How would I get in contact with a doctor who could preform this? We live in Atlanta, GA. And how risky is it for baby’s health?
Thanks!
Hi, Katherine. Congratulations on your pregnancy. The type of ectodermal dysplasia discussed in this article is x-linked hypohidrotic ectodermal dysplasia or XLHED. It does not include cleft palate. However, there are other types of ectodermal dysplasia which can involve cleft palate. I’m not sure which type affects you? However, the in-utero treatment discussed only pertains to XLHED and not to any other type of ectodermal dysplasia. At this time, the treatment is not available yet. EspeRare plans to conduct a clinical trial involving a larger number of babies to further study the treatment. The only risk for this treatment is the same risk associated with any amniocentesis. If you have any questions, don’t hesitate to call the NFED office at 618-566-2020. We’d love to talk to you and explain things. Good luck. Jodi, NFED Director, Marketing and Communications
[…] Fuente: Prenatal Treatment Restores Sweating in XLHED. https://nfed.org/blog/prenatal-treatment-restores-sweating […]
Hi,
It is so refreshing and exciting to hear about research that can alleviate the symptoms of potential x linked ED cases. I was wondering if there is any research in sight for Moms to avoid passing on the gene to their babies altogether. Currently the only way to do that is through IVF, but I was wondering if perhaps one day, it would be possible to have babies naturally without having the chance of passing it on.
Hello. We are not aware of any research that would prevent a mom from passing on the gene.Jodi, Director, NFED Marketing and Communications
Congratulations to NFED and our colleagues whose persistent work made this progress possible. As the scientist who cloned the EDA gene back in 1996, I can only share the excitement with you all. We later reported in 2001 that the EDA gene produces a soluble protein that is expressed in the skin of human fetuses already at 20 weeks of gestation. That the therapy works, administered early enough, makes complete sense. This is great news indeed.
Is there an update and if patient has a fetus who can be affected at what gestational age does the amnio/protein infusion need to be done
Hi, Tamara. We hope to have an update from EspeRare very soon for you. Dr. Schenider has said that the treatment protocol calls for the pregnant mom to get the protein delivered via amniocentesis at 26 weeks into the pregnancy. We encourage you to call our office to discuss further. Please call us at 618-566-2020. Thanks! ~ Jodi, Director, Marketing and Communications, NFED
My son is 10 years old and he has this EDA1, I am happy to hear that but is there any treatment for his age now in 2022? At least to fix his sweating problem the rest we can do it like putting teeth and plant hair. Please inform if there is any treatment and where did you reach so far in your research.
Thank
Hi, Jacob! Thanks for your message. Unfortunately, at this time, there are no treatments to reverse the symptoms of ectodermal dysplasias outside of this one, which studies have found is only effective when given before birth. They did do studies and newborns and did not have success. We will continue to push research forward in hopes that some day, we may find better treatments and even cures for ectodermal dysplasias. We do have a cooling guide available on our site at: https://nfed.org/wp-content/uploads/2016/06/Cooling-Guide.pdf, which may have some tips for your son. We’re glad you are a part of a community, and we will keep working to push research forward! – Veronica